Project #3: Vibrational frequencies in transition metal –
dioxygen adducts
Comparison of vibrational frequencies obtained from experiment
and theoretical calculations can be used to identify the mode of dioxygen
coordination in 1:1 transition metal – O2 adducts. This approach has particular merits in those
cases when the most generally applied theoretical procedure of computing energies
for different geometries and assigning the experimental complex as the lowest
energy optimized structure proves problematic.
In the case of 1:1 adducts of CuI with dioxygen, singlet
states can exhibit high degrees of multideterminantal character, leading to
potentially unreliable results from density functional theory (DFT) and
necessitating the use of expensive higher-order methods (e.g. multireference
second-order perturbation theory – CASPT2) to obtain accurate energies. In other cases, the relative energy of η1
and η2 metal/O2 complexes may be computed to
be within the range of error expected for DFT calculations (~3 kcal/mol with
the B3LYP functional). A common
experimental approach of testing for splitting in ν(O–O) values for mixed-label
isotopomers has recently been shown also to be highly problematic for assigning
the O2 binding mode.
Specifically, the lack of such splitting was shown not to be
diagnostic for side-on versus end-on dioxygen coordination.
However, in order for comparison of experimentally
and theoretically determined vibrational frequencies to be meaningful, computed
vibrational frequencies must be both inexpensive and highly accurate. DFT methods would seem to fit this
prescription, as they can efficiently handle large models and generate highly
reliable optimized geometries. However,
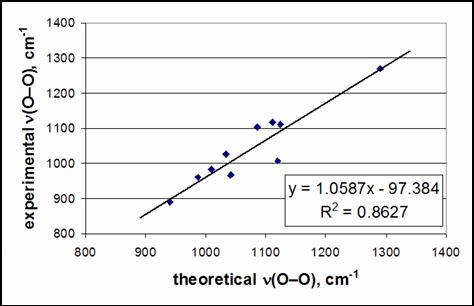
a body of evidence exists which suggests that ν(O–O) and ν(M–O)
in transition metal complexes computed by DFT methods are of insufficient
quality for the task at hand. For
example, computed O–O stretch frequencies (even when scaled) differ from
experimental values by 50 cm-1 in the side-on 1:1 Cu-O2
adducts supported by β-diketiminate and anilido-imine ligands when B3LYP
with a triple-zeta polarized basis set is used.
For other metal-O2 1:1 adducts, errors can routinely be in
the range of 20-50 cm-1. The
inaccuracy of computed frequencies in 1:1 M-O2 adducts is
highlighted by their poor correlation with experimental values (see
Figure). Known vibrational frequency
scaling factors for particular combinations of functionals and basis sets may
not be applicable to these cases since they were derived from studies on molecules
not containing transition metals.
This facet of research will therefore focus on determining an optimal
DFT-based method for computing ν(O–O) and ν(M–O) in transition metal
complexes. Combination(s) of density
functionals, basis sets, and scaling factors which lead to minimal error when
measured against experimental data will be ascertained. Pure, hybrid, and meta-GGA density
functionals will be considered, as will double- and triple-zeta basis sets with
and without polarization and diffuse functions.
A database of experimental ν(O–O) and ν(M–O) in 1:1 M-O2
adducts will be assembled in order to assess the accuracy of the DFT
methods. Isotopologue splitting
Δν(18O2) can also be included in
experimental/theoretical comparisons for cases where such experimental data are
available.
(1) Bauschlicher,
C. W.; Partridge, H. J. Chem. Phys. 1995, 103, 1788-1791.
(2) Cramer,
C. J.; Tolman, W. B.; Theopold, K. H.; Rheingold, A. L. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3635-3640.